Dando continuidade à série sobre Machine Learning, esse texto se concentra em abordar as ferramentas de deep learning que são utilizadas para a predição de estruturas tridimensionais de proteínas. Além disso, o recentemente lançado, AlphaFold, será nosso principal exemplo de aplicação. Mas antes de tudo, a questão fundamental é: por que, ainda hoje, há tantas incertezas e impasses com as aplicações para predição e modelagem de estruturas de proteínas?
Enovelamento proteico – uma dança complexa
Antes de abordarmos porque o enovelamento de proteínas é um problema, precisamos entender o que é uma proteína e o que é o seu enovelamento. As proteínas são polímeros de aminoácidos unidos através de ligações peptídicas, como se fossem as pérolas de um colar unidos pela corrente, que apresentam uma estrutura tridimensional intimamente relacionada à sua função. Elas estão entre as moléculas orgânicas mais abundantes nos sistemas biológicos e desempenham funções intrínsecas à vida, como por exemplo, as enzimas (um tipo de proteína) que participam da digestão dos alimentos.
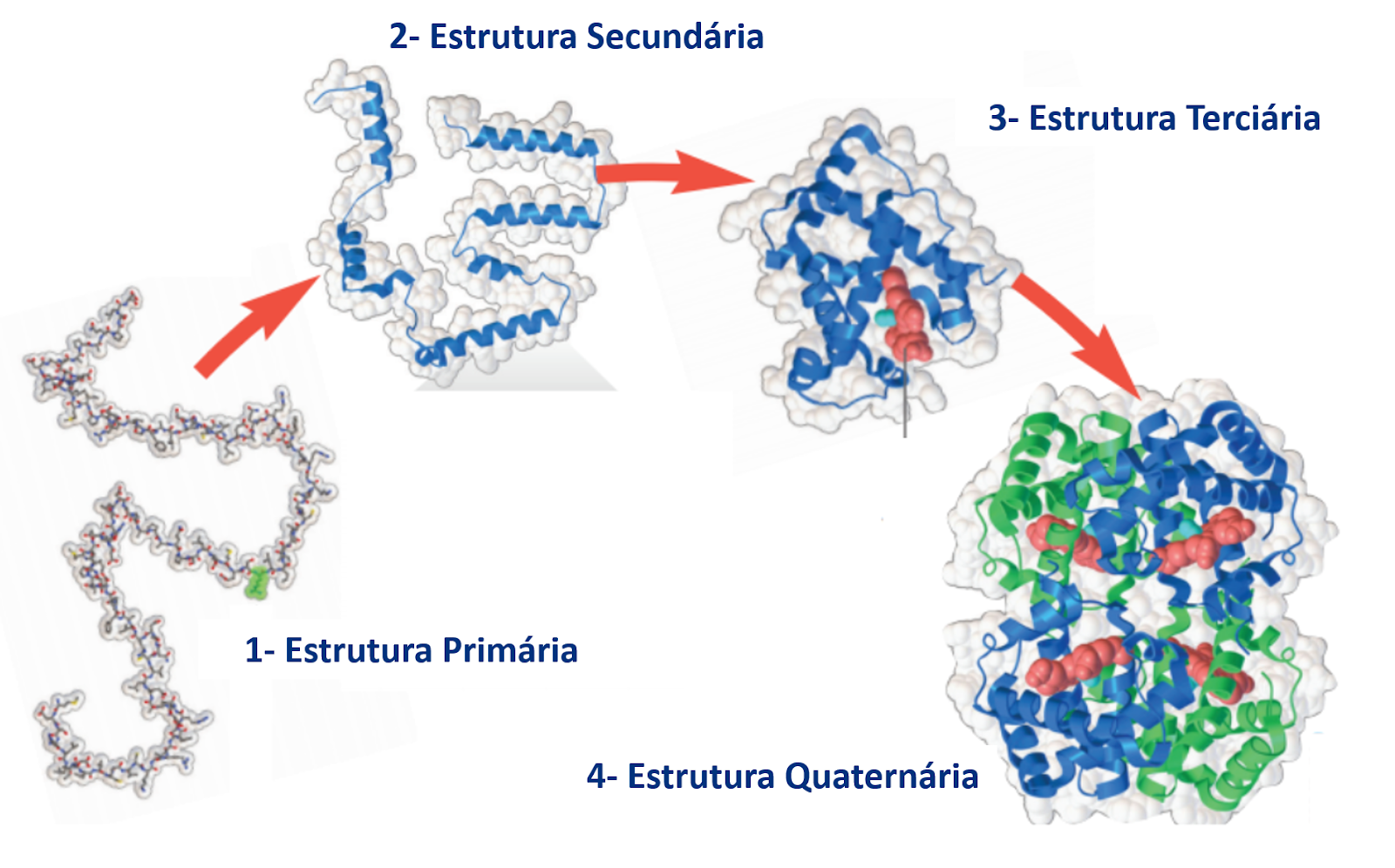
A primeira versão de uma proteína obtida através dos mecanismos do dogma central da biologia molecular e é a sequência de aminoácidos unidos pelas ligações peptídicas. Essa é chamada de estrutura primária. Porém, para atingir a sua estrutura tridimensional e consequente estrutura ou conformação nativa (aquela em que irá desempenhar sua função), a estrutura primária precisa passar pelo processo de enovelamento protéico.
O enovelamento é o processo pelo qual a estrutura primária da proteína “dobra-se sobre si mesmo”, dando origem às estruturas secundárias e terciárias através de interações químicas entre os átomos dos aminoácidos. A estrutura terciária é conformação tridimensional nativa de muitas proteínas, porém o arranjo entre estruturas terciárias pode dar origem à estrutura quaternária (entenda mais sobre as estruturas de proteínas neste link).
Em retrospectiva, existe um código genético específico para cada proteína, que é transcrito na forma de um RNA mensageiro, que é traduzido para uma estrutura primária proteica e que irá se modificar até atingir a conformação nativa. Parece bem estabelecido. Então, qual é o problema do enovelamento protéico?
A discussão desse problema é clichê, mas continua bastante atual e pode ser descrita com o Paradoxo de Levinthal. Cyrus Levinthal publicou em 1969 o seu experimento mental no qual, assumindo que cada aminoácido em uma ligação peptídica pode assumir dois ângulos diferentes (phi e psi), uma proteína com 300 aminoácidos possui 3300 (isso mesmo, um 3 seguido de 300 zeros) estruturas possíveis (partindo da probabilidade estatística) e, se a busca da conformação fosse aleatória, a proteína levaria em torno de 10129 anos para alcançar à estrutura nativa . O que é incompatível com a vida e sabemos que, na vida real, a conformação nativa é alcançada em segundos ou minutos.
Então, assumimos que as informações referentes às coordenações que os aminoácidos devem assumir no enovelamento proteico são transmitidas através do código genético para a sequência de aminoácidos (também conhecido como o dogma de Anfinsen). Porém, descobrir essas informações é um desafio complexo, o chamado “problema do enovelamento protéico” e, até novembro de 2020, a comunidade científica não tinha uma solução satisfatória para essa questão. Até o AlphaFold ser apresentado ao mundo.
A Bioinformática fornece métodos de contornar o paradoxo de Levinthal
Predizer a estrutura terciária e, consequentemente a conformação nativa, de uma proteína a partir apenas da sua sequência (estrutura primária) é um grande desafio. Obter a estrutura terciária de forma experimental, na bancada, através das metodologias de cristalografia também é um desafio, uma vez que é um processo trabalhoso e oneroso.
Porém, desde o final do século XX, métodos computacionais para realizar a predição da estrutura protéica tridimensional a partir de sua sequência de aminoácidos foram criados e aperfeiçoados graças à disponibilidade de informações e estruturas nos bancos de dados, como o Protein Data Bank (PDB) e o Universal Protein (UniProt). Existem dois métodos bastante utilizados: os baseados em homologia (template-based) e os baseados no conhecimento termodinâmico dos aminoácidos (template-free).
Os métodos baseados em homologia se dividem em modelagem comparativa e threading. Eles são ditos métodos dependentes de molde uma vez que parte do princípio de que a estrutura tridimensional de uma proteína (e consequente função) se mantém conservada ao longo da evolução. Logo, sequências semelhantes se enovelam em estruturas semelhantes ou idênticas. Na modelagem comparativa, a sequência-molde a sequência-alvo devem ser alinhadas e apresentar uma identidade mínima entre 25% e 30%. Já a modelagem por threading é utilizada para modelar estruturas que compartilham baixo grau de similaridade, mas que o enovelamento é similar ao de estruturas conhecidas. Para que isso seja possível, a sequência-alvo é fragmentada em busca por homólogos estruturais passando por diversos alinhamentos.
Os métodos de modelagem independente de molde são conhecidos por de novo e ab initio. Eles são utilizados em casos em que a sequência-alvo não compartilha identidade e similaridade com proteínas conhecidas. Porém esse tipo de modelagem é menos confiável que a modelagem por homologia e apresenta restrições quanto ao tamanho de sequências, sendo utilizadas para proteínas pequenas (até 200 aminoácidos). No método de novo são utilizadas informações estruturais e fragmentadas de bancos de dados para orientar o enovelamento. No método ab initio, não há o uso de informações de bancos de dados, apenas metodologias matemáticas e estatísticas para determinar as características termodinâmicas e obter o enovelamento.
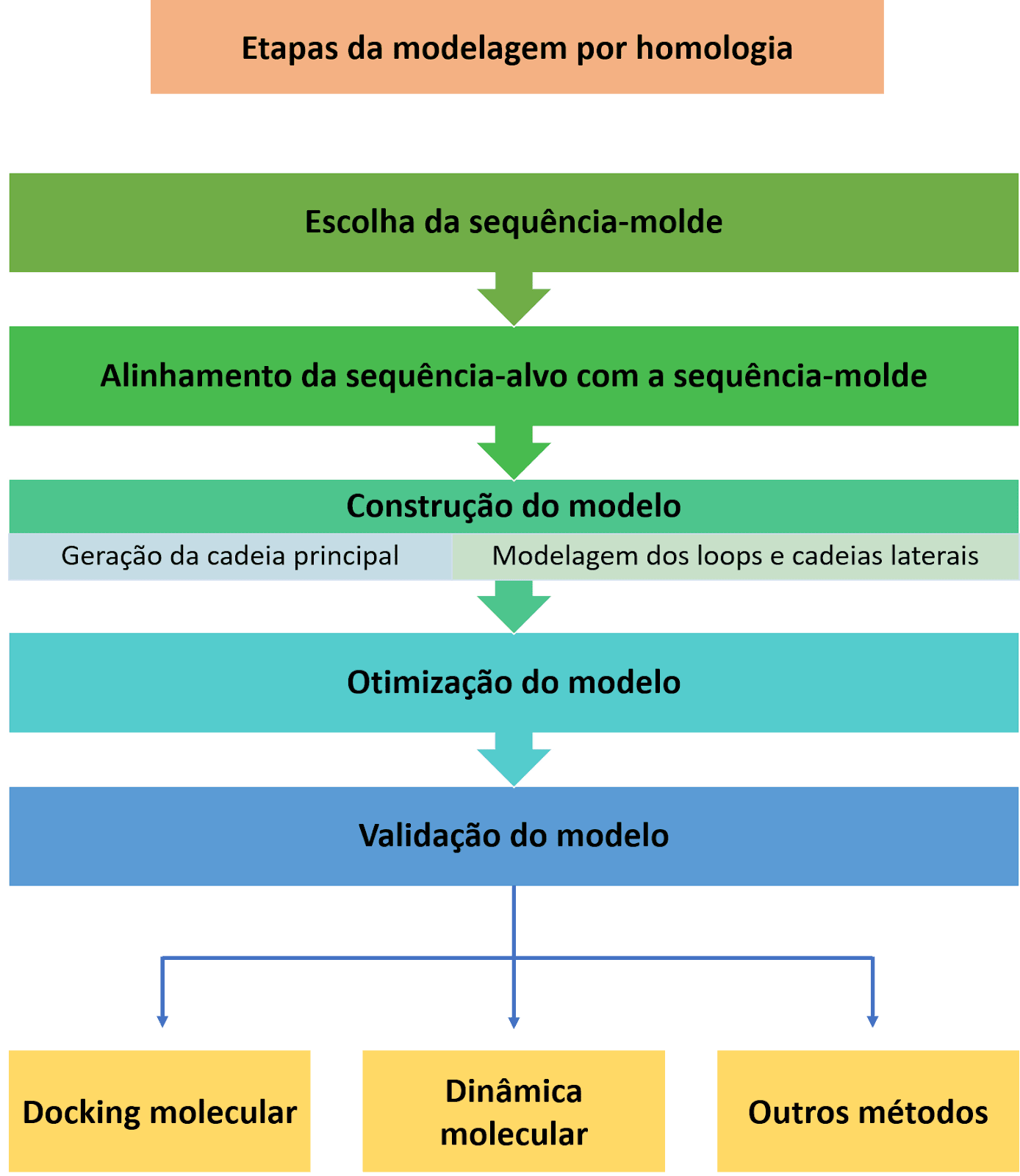
Por último, é importante destacar que os modelos gerados em ambos métodos precisam passar por avaliações de validação e otimização das estruturas. Essas ações podem já estar incluídas nos softwares de modelagem, bem como serem realizadas em outros servidores. Cada metodologia envolvida nessas etapas possui sistema e indicadores próprios para definir se cada modelo
O avanço tecnológico chamado AlphaFold
Todos os problemas previamente citados aparentemente se encontram solucionados com o lançamento do AlphaFold, mais especificamente a versão AlphaFold2 de dezembro de 2020. O AlphaFold foi produzido pela empresa DeepMind e é um sistema de inteligência artificial altamente inovativo que prediz a estrutura tridimensional de proteínas a partir da sua sequência de aminoácidos com grande acurácia e qualidade. Além disso, outro fato importante é que essa predição ocorre em minutos .
A arquitetura de software (organização de componentes e de interação com outros sistemas) utilizada pelos cientistas da DeepMind consiste em dois principais métodos para a obtenção das estruturas tridimensionais: o primeiro utilizada uma rede neural profunda e o segundo método utiliza o método matemático de gradiente descendente para otimizar a função de pontuação (scores) e a acurácia das estruturas. A seguir, iremos explorar os mecanismos por detrás da rede neural utilizada.
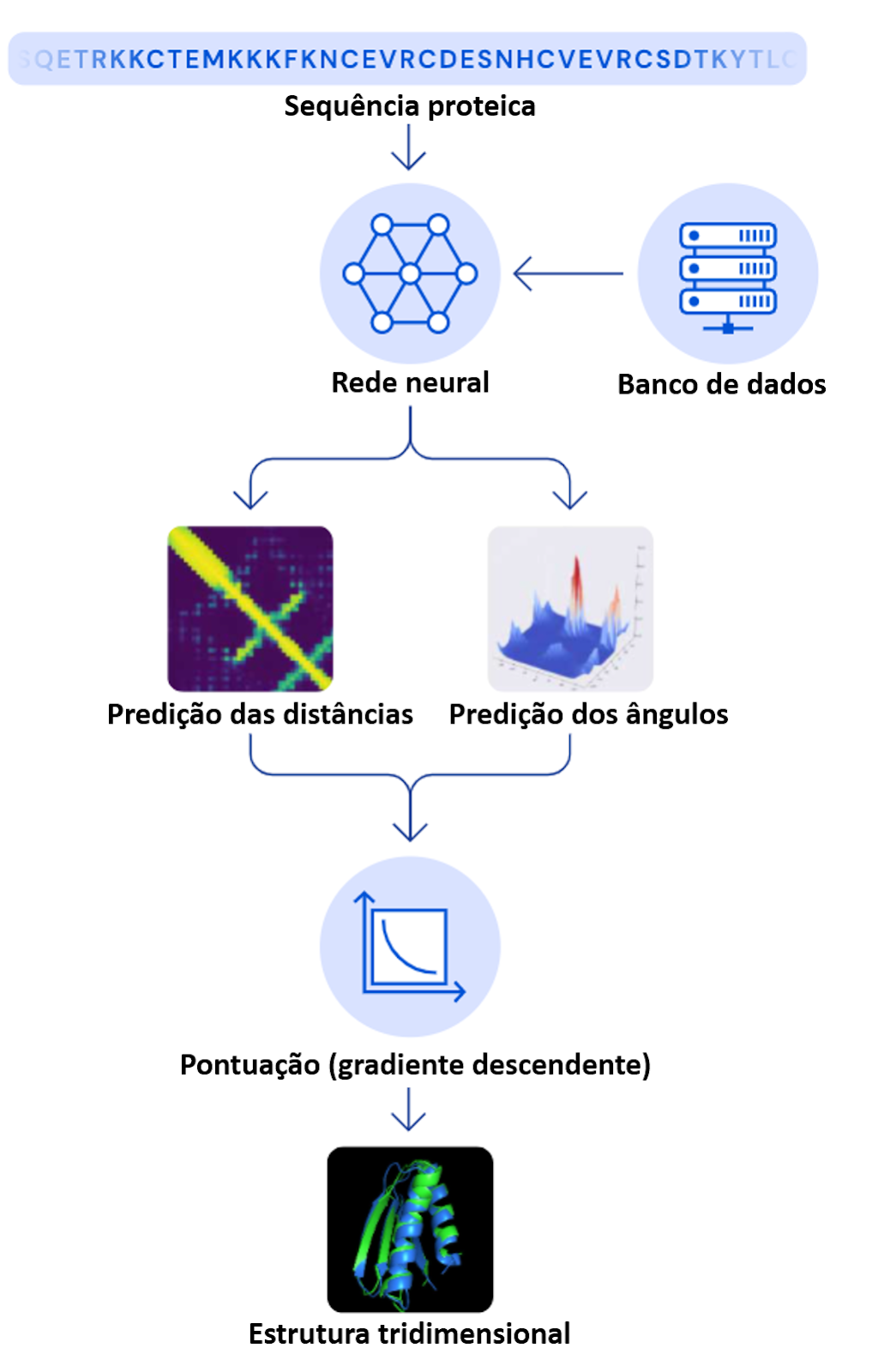
A rede neural do AlphaFold foi baseada na abordagem de aprendizado profundo (DL) chamada de Transformer. Porém, o time da DeepMind criou um novo tipo de transformer para trabalhar especificamente com estruturas tridimensionais, que eles denominaram de Invariant Point Attention (IPA). O modelo transformer utiliza o mecanismo de atenção (para informações mais técnicas, leia o artigo de lançamento do mecanismo de atenção), que é uma técnica para calcular a soma ponderada de valores, de forma que cada valor do input receba “atenção” e um peso de acordo com seu valor.
O transformer (e, consequentemente, o mecanismo de atenção) é bastante utilizado na tradução por máquina (machine translation), geração e leitura de documentos e na análise de sequências biológicas. Isso porque é um modelo bastante eficiente no processamento de dados de texto, possuindo a capacidade de aprender a relação entre as entidades distantes, assim como os aminoácidos distantes em uma sequência proteica se relacionam durante o enovelamento proteico.
Para conseguir gerar um modelo tridimensional a partir da sequência de aminoácidos, o AlphaFold utiliza informações de diversas bases de dados, como o PDB e o Uniprot. Dentre as informações captadas estão o alinhamento entre estruturas, características físico-químicas dos aminoácidos, características químicas das ligações e também informações da sequência genética da proteína-alvo.
A forma como o IPA foi desenvolvido permite a maximização do fluxo de informação em cada etapa, ou seja, cada conjunto de dados avaliados seguem e retornam entre todos os componentes do IPA. Isso garante que a estrutura final da proteína seja cada vez mais precisa. Além da flexibilidade entre os componentes do AlphaFold, a estrutura final obtida passa por todo o processo três vezes na etapa de reciclagem, permitindo a elevada acurácia na estrutura. Leia este artigo para compreender melhor o passo a passo e todos os componentes do AlphaFold.
O AlphaFold gera uma estrutura tridimensional com uma acurácia média de 95% quando comparado à estrutura experimental (obtida através métodos de cristalografia). Devido à sua acurácia e confiabilidade, ele foi utilizado para realizar a predição do proteoma humano, que antes possuía estruturas tridimensionais determinadas experimentalmente para apenas 17% das proteínas totais do proteoma. Com uso do AlphaFold, a cobertura passou a ser de 98,5% do proteoma determinado por simulação.
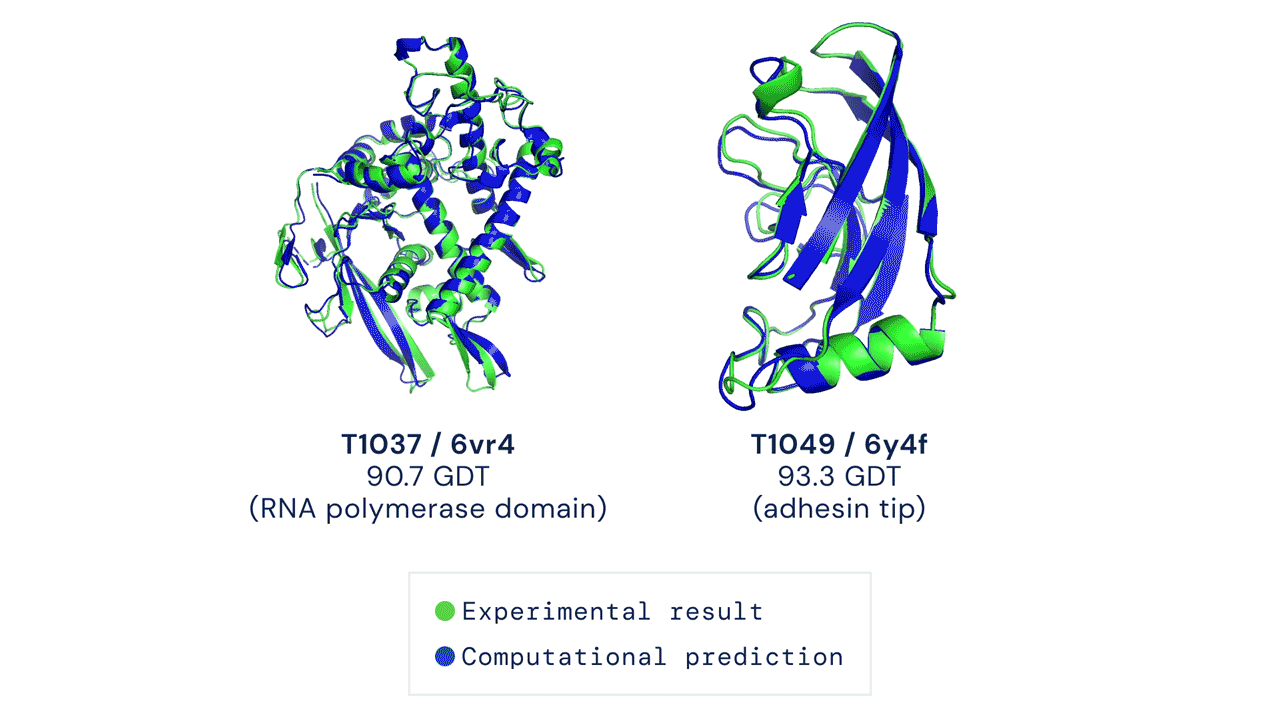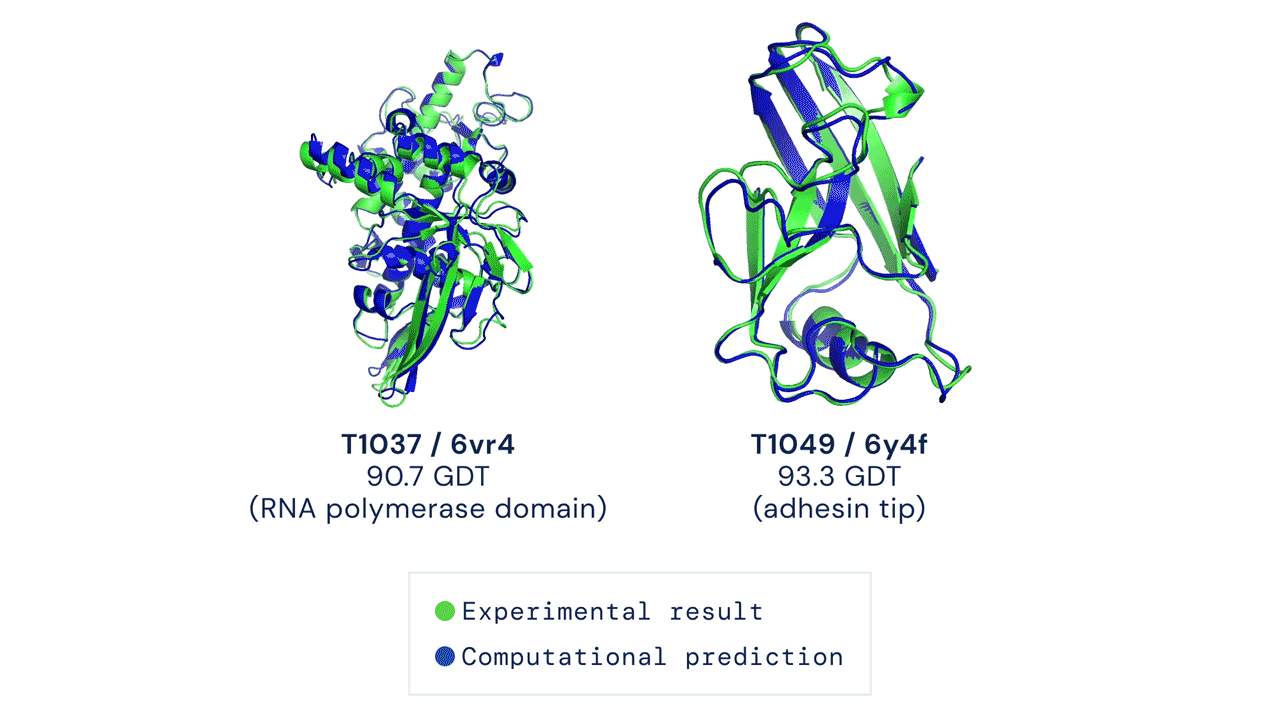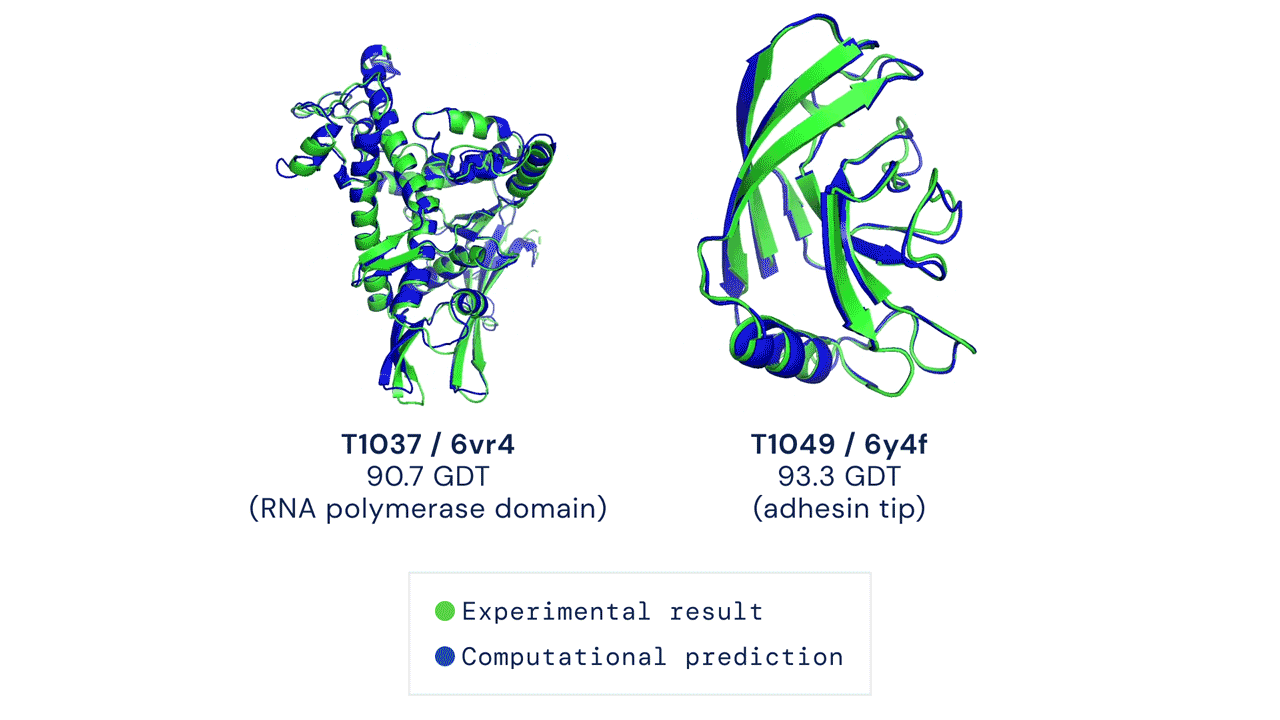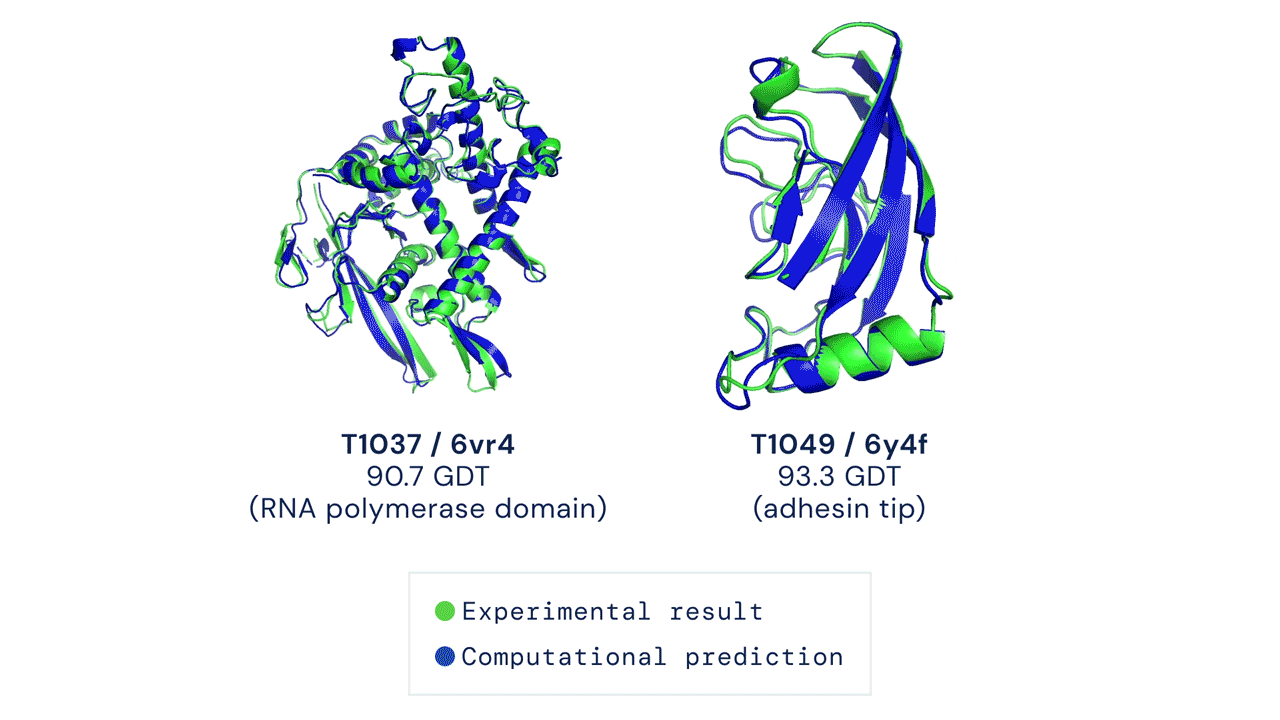
Porém, nem tudo são flores! O AlphaFold, apesar de ser um grande avanço e de apresentar uma acurácia média de 95% na modelagem das proteínas, ainda não resolve todos os problemas na área. Existem muitas estruturas ou partes de estruturas que têm acurácia bem baixa (valor indicado pelo próprio score do AlphaFold). Além disso, ele também não consegue prever diferentes conformações, por exemplo quando uma proteína está em um complexo.
Por enquanto, o AlphaFold realiza a predição de proteínas com até dois domínios, enquanto que na natureza a maioria das proteínas é multidomínio. Porém, essas e outras melhorias já estão sendo planejadas para implementação futura. Além disso, devido à importância do acesso aberto, a DeepMind em conjunto com o European Bioinformatics Institute (EMBL-EBI) desenvolveram o AlphaFold Protein Structure Database (AlphaFold DB), disponível para qualquer um que tenha interesse. O código fonte do AlphaFold também está disponível no GitHub e no Colab notebook para a predição de estruturas em computadores pessoais.
Em síntese, a inteligência artificial através do deep learning presente no AlphaFold realizou uma grande revolução na comunidade científica e irá auxiliar no desenvolvimento de pesquisas nas áreas que dependem de estruturas de proteínas. Apesar das presentes limitações, futuras implementações estão sendo desenvolvidas e serão de grande utilidade e, provavelmente, mais revoluções na ciência.

Cite este artigo:
LOURENÇO, D. A. Deep Learning na predição de estruturas proteicas – a revolução AlphaFold. Revista Blog do Profissão Biotec. V. 10, 2023. Disponível em: <https://profissaobiotec.com.br/deep-learning-predicao-estruturas-proteicas-revolucao-alphafold>. Acesso em: dd/mm/aaaa;
Referências
BITTENCOURT JUNIOR, J. A. Mecanismos de atenção. Setembro de 2018. Apresentação de Power Point. Disponível em: <https://ww2.inf.ufg.br/~anderson/deeplearning/20181/mecanismos_de_atencao_redes_neurais_profundas_deep_learning.pdf>. Acesso em: 17 Nov 2021.
DANTAS, D. Pay attention – Explicando o mecanismo de atenção. LAMFO. Disponível em: <https://lamfo-unb.github.io/2019/05/01/Pay-attention-Explicando-o-mecanismo-de-Atencao/>. Acesso em: 17 Nov 2021.
DENG, H., JIA, Y., ZHANG, Y. Protein structure prediction. Int J Mod Phys B., 32 (18): 1-18, 2018.
HADDAD, Y., ADAM, V., HEGER, Z. Ten quick tips for homology modeling of high-resolution protein 3D structures. PLOS Computational Biology, 16 (4): e1007449, 2020. DOI: https://doi.org/10.1371/journal.pcbi.1007449.
MARTÍNEZ, L. O paradoxo do enovelamento de proteínas: um modelo simples. 26 de Outubro de 2016. Apresentação de Power Point. Disponível em: <http://leandro.iqm.unicamp.br/leandro/shtml/folding_slides.pdf>. Acesso em: 12 Nov. 2021.
MAXIME. What is a Transformer?. Inside Machine Learning. Disponível em: <https://medium.com/inside-machine-learning/what-is-a-transformer-d07dd1fbec04>. Acesso em: 17 Nov 2021.
NEIS, A. Conceitos Básicos em Modelagem de Proteínas. Omixdata;. Disponível em: <https://medium.com/omixdata/conceitos-b%C3%A1sicos-em-modelagem-de-prote%C3%ADnas-b9f8ac2c0b84>. Acesso em: 15 Nov. 2021.
NELSON, D. L., COX, M. M. Princípios de Bioquímica de Lehninger. 7ª Ed. Porto Alegre: Artmed, 2018.
SENIOR, A., JUMPER, J., HASSABIS, D., KOHLI, P. AlphaFold: Using AI for scientific discovery. DeepMind. Disponível em: <https://deepmind.com/blog/article/AlphaFold-Using-AI-for-scientific-discovery>. Acesso em: 17 Nov 2021.
SILVA, L. X., BASTOS, L. L., SANTOS, L. H. Modelagem computacional de proteínas. In: BIOINFO – Revista Brasileira de Bioinformática e Biologia Computacional. 1. Ed. Vol. 1. Lagoa Santa: Editora Alfahelix, 2021. DOI: 10.51780/978-6-599-275326.
TOEWS, R. AlphaFold is the most important achievement in AI – Ever. Forbes. Disponível em: <https://www.forbes.com/sites/robtoews/2021/10/03/alphafold-is-the-most-important-achievement-in-ai-ever/?sh=55e4db156e0a>. Acesso em: 17 Nov 2021.
Fonte da imagem destacada: NIH – Visuals online.