

Pesquisadores do mundo todo têm reunido esforços em uma corrida contra o tempo na procura por medicamentos potencialmente eficazes contra o SARS-CoV-2. Por conta disso, tem se tornado cada vez mais comum a busca pela utilização de novas tecnologias e abordagens metodológicas que possam agilizar esse processo. No entanto, como se pode acelerar a busca por novos fármacos? O custo dessa busca é elevado?
A busca por novos fármacos pode ser acelerada através de metodologias in silico, ou seja, totalmente computacionais, que são conhecidos por sua rapidez e baixo custo, permitindo a análise de uma grande quantidade de dados como, por exemplo, milhares de possíveis medicamentos contra o SARS-CoV-2, em poucas semanas. A Ancoragem Molecular, do inglês Molecular Docking, é a exemplificação de um dos grandes aliados nessa procura, permitindo a avaliação da interação entre proteína viral e ligante, ou seja, vírus e possível medicamento.
Vírus e medicamento interagindo virtualmente? Como isso é possível?
O Molecular Docking é um método utilizado como uma primeira triagem, ou um ponto de partida, no estudo do potencial farmacêutico e/ou medicinal de um composto ou até mesmo famílias inteiras de compostos, podendo ser feito o uso de bibliotecas e repositórios de estruturas de medicamentos. Os principais alvos de estudo contra o SARS-CoV-2 tem sido medicamentos já testados, seguros, e com eficácia comprovada contra determinadas doenças, sendo esse método denominado reposicionamento de fármacos.
A principal vantagem em se utilizar esse tipo de abordagem consiste em já se ter o conhecimento prévio da eficácia, assim como da toxicidade e dos efeitos colaterais de medicamentos já testados e conhecidos, as quais são informações que se tem apenas através de testes clínicos, agilizando e tornando mais barato o processo na busca por um medicamento eficaz contra o SARS-CoV-2.
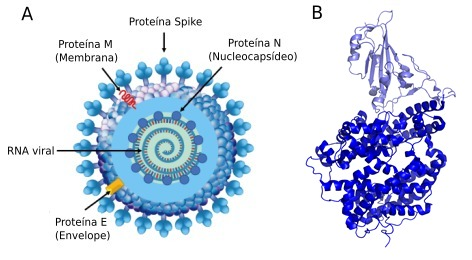
Na estrutura do SARS-CoV-2 se observa a presença da proteína spike, a qual confere a esse vírus a sua “coroa”. Essa proteína é a responsável por facilitar a entrada do vírus nas células humanas e tem sido objeto de muitos estudos de molecular docking. A abordagem proposta por essa metodologia permite um estudo detalhado da interação entre uma pequena molécula (o medicamento em estudo) e o sítio de ligação de uma proteína alvo.
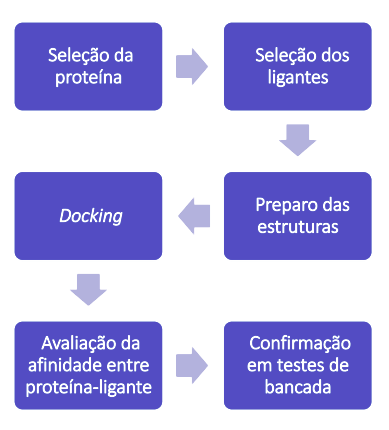
Portanto, como é observado se houve interação entre o composto testado e a proteína alvo? Como determinar se o procedimento de docking realizado pode ser considerado satisfatório? Para isso, se tem a divisão do método em três principais etapas: primeiramente, um estudo prévio sobre a conformação do composto, sua orientação e localização, após é feita a análise e preparo da estrutura da proteína, incluindo o estudo dos prováveis sítios de ligação e, como última etapa, é feito o molecular docking propriamente dito, com a avaliação da afinidade da ligação entre proteína e medicamento.
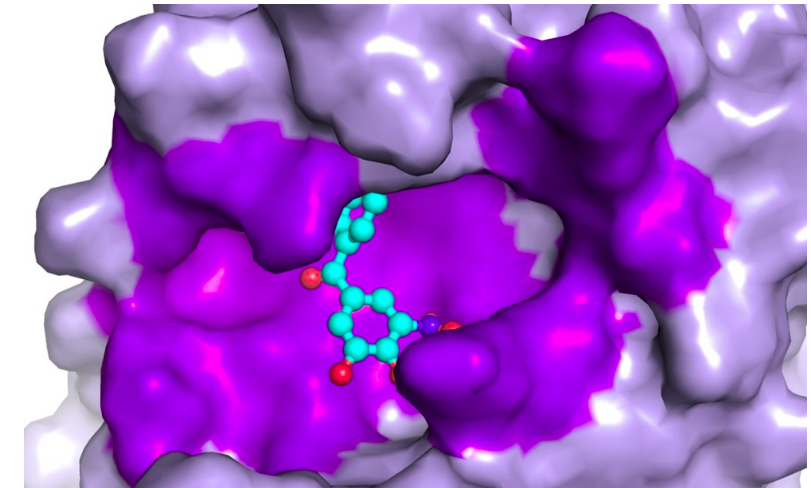
Um fator que pode ser determinante na qualidade dos resultados de um docking consiste em se saber previamente a localização do potencial sítio de ligação da proteína que irá receber o ligante. Caso o sítio de ligação ainda seja desconhecido, uma abordagem a ser considerada é a comparação da proteína e da molécula em estudo com alguma proteína que apresenta estrutura ou função semelhante e que, em alguns casos, é encontrada em bancos como o Protein Data Bank já complexada com alguma molécula de tamanho similar ao medicamento em estudo, o que fornece uma noção de possível sítio de ligação do complexo proteína-ligante que se quer estudar.
Quando não se tem nenhum conhecimento prévio sobre o sítio de ligação, pode ser feita a utilização de programas que ajudem nessa predição como, por exemplo, o KVFinder, uma ferramenta ágil e versátil, desenvolvida aqui no Brasil por pesquisadores do Laboratório Nacional de Biociências (LNBio), que auxilia na busca pelas cavidades de uma proteína que possam ser potenciais sítios de ligação, estando disponíveis no site do KVFinder vídeos contendo tutoriais e link para download. Ainda há os casos em que se realiza o encaixe proteína-ligante sem nenhum conhecimento sobre o sítio de ligação, sendo denominado de docking cego.
Dentre as ferramentas que podem ser utilizadas para a realização de experimentos de docking, o DockThor configura o exemplo de uma ótima ferramenta, desenvolvida por pesquisadores brasileiros do Laboratório Nacional de Computação Científica (LNCC). Essa ferramenta pode ser encontrada no portal DockThor, no qual também são disponibilizadas aos usuários, estruturas completas das proteínas que compõem o vírus SARS-CoV-2, com potenciais alvos para triagens virtuais de fármacos.
E o que acontece após essa triagem virtual de medicamentos? Qual é o próximo passo? Geralmente são testadas milhares de moléculas contra a proteína viral alvo. No entanto, dentre essas milhares, apenas algumas que demonstraram maior afinidade, ou seja, se ligaram de forma mais eficaz à proteína do vírus, irão para o próximo passo, que será os testes, ou ensaios clínicos de fato.
Resultados brasileiros de molecular docking contra o SARS-CoV-2
Assim como no desenvolvimento de ferramentas que tem facilitado, e muito, o processo de busca por medicamentos contra o SARS-CoV-2, os pesquisadores brasileiros têm se destacado também através de resultados bastante expressivos nessa corrida por fármacos.
Pesquisadores do Laboratório Nacional de Biociências do Centro Nacional de Pesquisa em Energia e Materiais (LNBio-CNPEM), localizado em Campinas/SP, desenvolveram pesquisa baseada no reposicionamento de fármacos, na qual foram testados 2 mil medicamentos já autorizados pelo Food and Drug Administration (FDA), resultando em 16 medicamentos com potencial de inibição do vírus.
O conhecimento, não somente da estrutura da proteína spike, mas também das demais estruturas protéicas do vírus, incluindo a proteína de membrana (M), abriu um leque de possibilidades no que diz respeito ao molecular docking.
E foi através da elucidação da estrutura da proteína M, que pesquisadoras da Fiocruz em Curitiba/PR, ao rastrear 4.334 compostos, evidenciaram o potencial de modificadores de coagulação no tratamento da COVID-19, a partir da observação da semelhança das estruturas da proteína M do SARS-CoV-2 e células humanas envolvidas nos processos de coagulação, denominadas trombina e Fator Xa.
Os desafios e perspectivas na obtenção de novos fármacos
A obtenção de novos fármacos, desde a pesquisa até seu desenvolvimento, é geralmente um processo oneroso, demorado e bastante complexo. Mas, graças ao progresso da biotecnologia, na chamada Revolução Biotecnológica, – que inclui a ascensão de áreas como a genômica, proteômica e a metabolômica -, estão disponíveis atualmente informações que podem ser fundamentais na otimização da descoberta de novos fármacos. Dentre essas informações, configuram exemplos importantes os dados de expressão gênica, a elucidação de estruturas e funções de proteínas, assim como as descrições acerca do metabolismo de células e tecidos.
Mesmo com o aumento significativo de informações úteis ao processo de obtenção de fármacos, um dos maiores desafios da química medicinal ainda consiste na capacidade de inovar no que diz respeito ao descobrimento de compostos químicos que apresentem potencial medicinal. Dessa forma, se tem apostado na junção entre métodos experimentais e computacionais, a fim de promover o aumento na probabilidade da descoberta de compostos bioativos a partir do estudo de bibliotecas de compostos químicos disponíveis experimentalmente e/ou virtualmente.

Cite este artigo:
GODOY, B. R. B. Molecular Docking: o aliado na busca por medicamentos contra o SARS-CoV-2. Revista Blog do Profissão Biotec, v. 8, 2021. Disponível em:<https://profissaobiotec.com.br/molecular-docking-medicamentos-contra-sars-cov-2/>. Acesso: dd/mm/aaaa.



